Advanced FASTA Sequence Manipulation
GenBeans offers several tools & functions for an advanced manipulation of FASTA files.
![]() Always save a copy of your original FASTA file before making advanced modifications.
Always save a copy of your original FASTA file before making advanced modifications.
Accessing Advanced FASTA tools
1. Open the FASTA file in Explorer.
2. Select the main FASTA sequence node in the Explorer.
3. Advanced FASTA functions are found in the main menu under the Tools item:

Order FASTA sequences functions
These two tools order sequences alphabetically based on either the sequence name or the sequence accession number. If the identifier contains multiple delimiters and you do not know which field will be used as name or accession, use the Customize FASTA Header function to isolate them. If a sequence with no name or no accession is encounter, the method is aborted and the file remains unchanged.
Customize FASTA Header function
The description line in FASTA format is often used to pack information related to the database the sequences are originating from. The following table shows a few examples of database identifiers found in FASTA definition lines (source: The NCBI Handbook):
Database name | Identifier syntax | |
GenBank |
gi|gi-number|gb|accession|locus |
|
EMBL Data Library |
gi|gi-number|emb|accession|locus |
|
DDBJ, DNA Database of Japan |
gi|gi-number|dbj|accession|locus |
|
NBRF PIR |
pir||entry |
|
Protein Research Foundation |
prf||name |
|
SWISS-PROT |
sp|accession|name |
|
Brookhaven Protein Data Bank |
pdb|entry|chain |
|
Patents |
pat|country|number |
|
General database identifier |
gnl|database|identifier |
|
NCBI Reference Sequence |
ref|accession|locus |
|
Local Sequence identifier |
lcl|identifier |
|
|
|
The FASTA Header Customizer can be used to remap fields in the definition line; after clicking the OK button, fields are isolated by the input header divider, numbered according to their occurence in the definition line and remapped according to the new field mapping; up to 10 fieds can be remapped and will be separated using the output field divider.
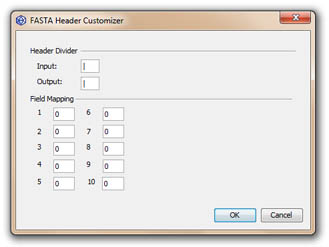
For example, customizing a FASTA file originating from the GenBank database with the syntax ">gi|gi-number|gb|accession|locus" using '|' as input delimiter and a simple 1-5 mapping will change the syntax of the definition line to ">locus".
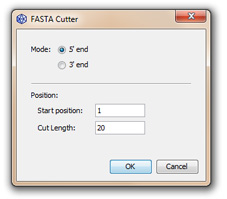
Truncate FASTA sequences
This function removes a few nucleotides to each sequence from the start position over the cut length. When operated from the 3' end, numbering is taken on the revese complement sequence. This tool works on protein sequences as well.
Extract FASTA DNA sequences
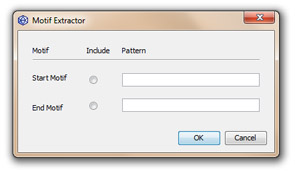
This tool extracts DNA sequences using IUPAC DNA motifs.
Remove duplicate FASTA sequences
This tool deleted sequences with duplicate name.
Remove selected FASTA sequences
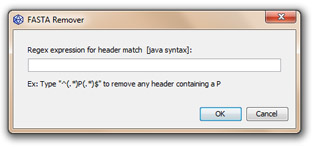
This tool removes sequences having a particular regex motif in their definition line.
![]() See Also
See Also