Working with FASTA Files
FASTA Format
The FASTA format is a simple text-based format for representing biological sequences. It is a multisequence format where each new sequence starts by a single description line followed by the sequence data as in the following example:
>M20573 Mouse Ig active kappa chain mRNA (V-J2), anti-lysozyme antibody TGCCAACAATGGGGTCGTAACCCCACGTTCGGAGGGGGGACCAAGCTGGAAATAAAA
1. Description line. The description line starts by the greater than symbol (">") and is followed by the name of the sequence and comments separated by a blank space from the name. In fact there is no strict convention for writing the description line and variations from the standard "><name> <description>" are frequent. GenBeans understands the description line as follows:
>gi|<identifier>|<namespace>|<accession>.<version>|<name> <description> >gi|<identifier>|<namespace>|<accession>|<name> <description> ><namespace>|<accession>.<version>|<name> <description> ><namespace>|<accession>|<name> <description> ><name> <description>
The description is optional in all cases; the version defaults to 0 if not provided and the namespace is the local space ("lcl") by default.
2. Sequence data. Sequences are represented in the standard one-letter IUB/IUPAC amino acid and nucleic acid codes. It is recommended that each line of data contains no more than 80 characters.
FASTA files and extensions
By default, files terminated by the extension "fasta", "faa", "fa", "fas", and "fsa" are recognized as FASTA files by the Explorer and displayed with a FASTA icon (![]() ). Associations between FASTA files and extensions can be adjusted in the Options > Miscellaneous > Files panel.
). Associations between FASTA files and extensions can be adjusted in the Options > Miscellaneous > Files panel.
Sequence recognition and sequence type
FASTA files are automatically recognized by GenBeans. First GenBeans tries to parse the sequence data as a protein, then, if it fails, as a DNA, and if it fails again, the type of the sequence is left to unknown. The user can choose or correct at any time the sequence type. Four types are possible: DNA (![]() ), RNA (
), RNA (![]() ), protein (
), protein (![]() ), and unknown (
), and unknown (![]() ). To change the type, either activate the contextual menu in the Explorer by right-clicking the file node and then select the new type in the Sequence Type submenu or double click the file node to open the file in the editor and click in the drop-down menu on the left side of the editor toolbar:
). To change the type, either activate the contextual menu in the Explorer by right-clicking the file node and then select the new type in the Sequence Type submenu or double click the file node to open the file in the editor and click in the drop-down menu on the left side of the editor toolbar:

GenBeans remembers between work sessions the type of the sequence and, therefore, you need to set the type only the first time your open your sequence in the editor.
Viewing FASTA sequences in the Explorer
Once a file is parsed and sequences are identified, you can view them by clicking in the '+' sign in front of the file node:
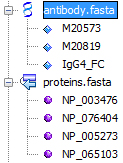
Sequences contained in the FASTA file appear after expanding the node in the Explorer. You can manipulate each sequence individually or in group by operating selection; you can perform drag & drop operations and copy & paste operations as well. For example, try to reorder the sequences in a single FASTAS file already opened in the Editor and look at the transformations while you drag a sequence up or down.
Contextual menu
You can access the contextual menu by right-clicking on the sequence node or the file node:

• Open: Open the FASTA file in the Editor at the first line of the sequence.
• Sequence Analysis: Access to varied sequence analyses such as translation, restriction analysis, etc.
• Export Sequence: Export to a new file in varied formats.
• Transform: Operate varied transformations on the sequences such as reverse complementation.
• Justify: Justify the sequences; the number of characters per line and the use of capital is set in the Options panel Biology > Editing > Sequence Editing.
The FASTA editor
Opening files in the Editor
To open a FASTA file, either double-clik on the corresponding node in the Explorer or choose the menu File > Open from the main menu to access the file dialog.
Parsing as you type
Once opened in the editor, the FASTA file is automatically parsed as you type; single sequences are recognized and description lines are highlighted appropriately. The color schema can be adjusted in the Fonts & Colors category of the Options panel. When an error in the FASTA format is encountered, a special red highlight is shown in the editor as in the following example:
![]()
A warning is emitted in some special cases, typically when a duplicate sequence name is found as in the following example:
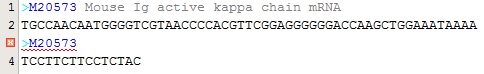
Sequence selection
Selections made inside the sequence data are recognized as subsequence selection. Information on the selected subsequences are found in the Properties Window or the status bar. This may come in very handy, for example, when searching for an oligonucleotide with a particular Tm. A contextual menu is accessed by right-clicking in the selection and offers a convenient way to analyze subsequences. Note that when no selection is made, the contextual menu applies for the entire sequence.
Editor toolbar
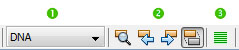
1. Dropdown menu to select the type of sequence between DNA, RNA, protein, and unknown. The chosen type will be remembered during the next GenBeans sessions.
2. Text Search inside the editor (Find selection, Find previous occurence, Find next occurence, Toggle highlight search). As of GenBeans 3.6.1 sequence search is not enabled in GenBeans.
3. Justify all sequences. Line width and use of capital letters to represent sequence symbols can be set in the Sequence Option Panel.
Keyboard shortcuts
Beside usual shorcuts, a few sequence specific shortcuts are defined:
Ctrl-T - Analyze 3x frame translations on nucleotidic sequences.
Ctrl-Shift-T - Analyze all 6x frame translations on nucleotidic sequences.
![]() See Also
See Also